Съдържание
- Причини
- Симптоми
- Изпити и тестове
- лечение
- Групи за подкрепа
- Кога да се свържете с медицински специалист
- Предотвратяване
- Алтернативни имена
- Снимки
- Препратки
- Дата на преразглеждане 8/6/2017
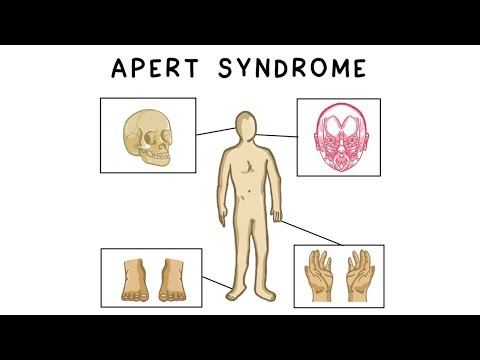
Синдромът на Apert е генетично заболяване, при което шевовете между костите на черепа се затварят по-рано от нормалното. Това влияе върху формата на главата и лицето.
Причини
Синдромът на Apert може да се предава чрез семейства (наследствени) като автозомно доминантна черта. Това означава, че само един родител трябва да премине погрешния ген, за да може детето да е в състояние.
Някои случаи могат да се появят без известна фамилна анамнеза.
Синдромът на Apert е причинен от една от двете промени в FGFR2 ген. Този ген дефект кара някои от костните конци на черепа да се затворят твърде рано. Това състояние се нарича craniosynostosis.
Симптоми
Симптомите включват:
- Ранно затваряне на шевовете между костите на черепа, отбелязано чрез подрязване по шевовете (краниосиностоза)
- Чести ушни инфекции
- Сливане или тежка лента на 2-ри, 3-ти и 4-ти пръст, често наричани "ръкавици с ръкавици"
- Загуба на слуха
- Голямо или късно затварящо меко петно върху черепа на бебето
- Възможно, бавно интелектуално развитие (варира от човек на човек)
- Видими или изпъкнали очи
- Тежко недоразвиване на средната повърхност
- Аномалии на скелета (крайниците)
- Къса височина
- Ленти или сливане на пръстите
Няколко други синдрома могат да доведат до сходен вид на лицето и главата, но не включват тежките характеристики на ръката и краката на синдрома на Apert. Тези подобни синдроми включват:
- Синдром на Карпентър (kleeblattschadel, деформиране на черепа на детелината)
- Болест на Crouzon (краниофациална дизостоза)
- Синдром на Pfeiffer
- Синдром на Saethre-Chotzen
Изпити и тестове
Доставчикът на здравни услуги ще извърши физически изпит. Ще бъдат направени рентгенови снимки на ръцете, краката и черепа. Винаги трябва да се извършват тестове за изслушване.
Генетичното изследване може да потвърди диагнозата на синдрома на Apert.
лечение
Лечението се състои от операция за коригиране на абнормния костен растеж на черепа, както и за сливане на пръстите на ръцете и краката. Децата с това разстройство трябва да бъдат прегледани от специализиран екип на черепно-лицевата хирургия в детски медицински център.
Ако има проблеми със слуха, трябва да се консултирате със специалист по слуха.
Групи за подкрепа
Детска краниофациална асоциация: www.ccakids.com
Кога да се свържете с медицински специалист
Обадете се на вашия доставчик, ако имате фамилна анамнеза за синдром на Apert или забележите, че черепът на вашето бебе не се развива нормално.
Предотвратяване
Генетичното консултиране може да бъде полезно, ако имате фамилна анамнеза за това заболяване и планирате да забременеете. Вашият доставчик може да тества вашето бебе за това заболяване по време на бременност.
Алтернативни имена
Acrocephalosyndactyly
Снимки

синдактилия
Препратки
Kinsman SL, Johnston MV. Вродени аномалии на централната нервна система. В: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Нелсън Учебник по педиатрия, 20-то издание. Филаделфия, Пенсилвания: Elsevier; 2016: глава 591.
Робин НХ, Фалк МЮ, Халдеман-Енглерт ЧР. FGFR-свързани craniosynostosis синдроми. GeneReviews, 2011: 11. PMID: 20301628 www.ncbi.nlm.nih.gov/pubmed/20301628.
Дата на преразглеждане 8/6/2017
Актуализиран от: Анна С. Еденс Хърст, доктор по медицина, асистент по медицинска генетика, Университетът в Алабама в Бирмингам, Бирмингам, Албания. Преглед, предоставен от VeriMed Healthcare Network. Също така бяха разгледани от Дейвид Зиве, MD, MHA, медицински директор, Brenda Conaway, редакционен директор и A.D.A.M. Редакционен екип.